Molecular bases of protein aggregation diseases
Protein aggregation has been linked to a range of human diseases of previously unknown nature. These are characterized by the accumulation of so-called amyloid structures formed by aggregation prone proteins such as α-synuclein (α-Syn) in Parkinson’s disease (PD). An amyloid is a highly ordered, fibrillar and polymeric protein structure with a unique set of biophysical characteristics, such as extreme protease resistance and stability, assembly by nucleated polymerization and the capability of specific self-templating.
Genetic screens in model organisms and genome-wide association studies (GWAS) have identified genes that reduce pathological effects of amyloidogenic proteins or that, in humans, affect the predisposition to the respective disease. Many of these genetic modulators are conserved from yeast to mammals, but their mechanisms of actions are unknown. Amyloid structures can also be released from a cell and “infect” neighboring cells, thus accelerating disease progression.
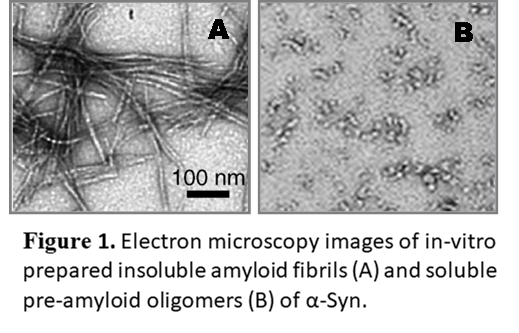
So far, amyloid structures and their assembly kinetics have been mainly characterized in vitro, using purified proteins forced to polymerize into a fibrillar state by specific buffer and incubation conditions. This considerably limits our current knowledge of amyloids and amyloidogenesis, since the conformations adopted by proteins in cells are regulated by multiple co-occurring events, specific to the cellular context - molecular crowding, binding of ligands, or post-translational modifications, for example. This has led to concerns about the validity of in vitro amyloid models, and resulted in an active debate on the structural states acquired by the model amyloidogenic protein α-Syn, a key player in PD.
Using a combination of structural and functional proteomic approaches and classical biochemical and molecular biology tools, our laboratory studies:
1. The cellular structures of amyloidogenic proteins under physiological conditions and upon development of neurodegenerative diseases
2. The effects of genetic and chemical modifers of the toxicity of amyloidogenic proteins
3. The trans-cellular spread of amyloids
Selected Publications
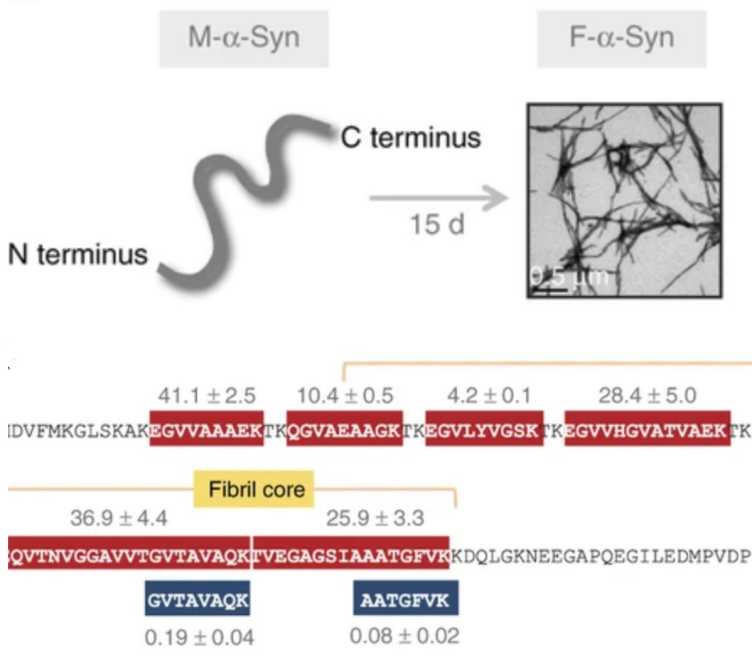
M.T. Mackmull, L. Nagel, F. Sesterhenn, J. Muntel, J. Grossbach, P. Stalder, R. Bruderer, L. Reiter, W.D.J. van de Berg, N. de Souza, A. Beyer, P. Picotti. external page Global, in situ analysis of the structural proteome in individuals with Parkinson's disease to identify a new class of biomarker. (2022) Nat. Struct. Mol. Biol., 29(10):978-989.

M. Soste, K. Charmpi, F. Lampert, J.A. Gerez, M. van Oostrum, L. Malinovska, P.J. Boersema, N.C. Prymaczok, R. Riek, M. Peter, S. Vanni, A. Beyer, P. Picotti. external page Proteomics-Based Monitoring of Pathway Activity Reveals that Blocking Diacylglycerol Biosynthesis Rescues from Alpha-Synuclein Toxiciexternal page ty. (2019) Cell Syst. 9(3):309-320.e8.
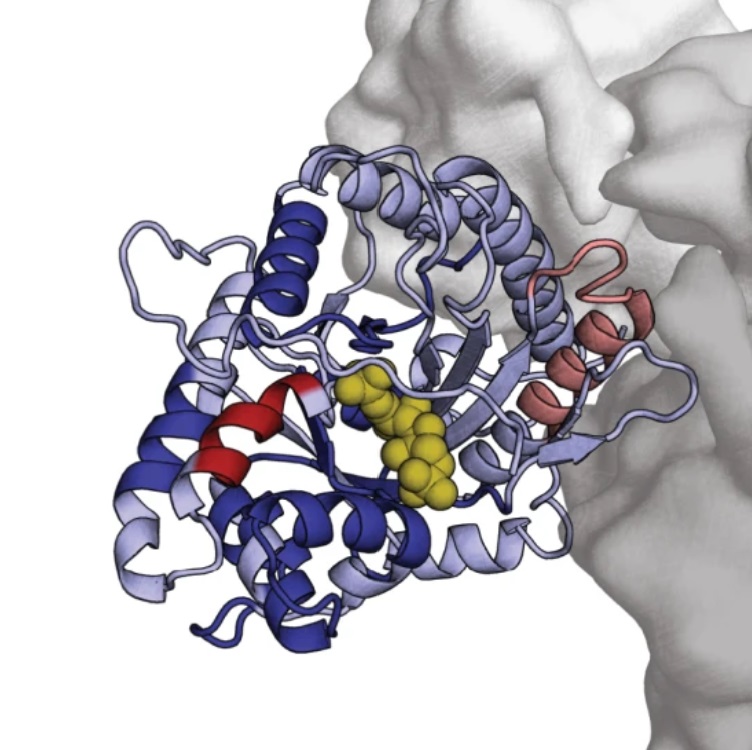
Y. Feng, G. De Franceschi, A. Kahraman, M. Soste, A. Melnik, P.J. Boersema, P.P. de Laureto, Y. Nikolaev, A.P. Oliveira, P. Picotti. external page Global analysis of protein structural changes in complex proteomes. (2014) Nat. Biotechnol., 32 (10), 1036-1044.

Gerez JA, Prymaczok NC, Rockenstein E, Herrmann US, Schwarz P, Adame A, Enchev RI, Courtheoux T, Boersema PJ, Riek R, Peter M, Aguzzi A, Masliah E, Picotti P. external page A cullin-RING ubiquitin ligase targets exogenous α-synuclein and inhibits Lewy body-like pathology. (2019) Sci Transl Med. 11(495):eaau6722.